Many eukaryotic microorganisms have a simple structure that does not lend itself well to the traditional, morphology-based, classification systems. This is often due to secondary loss of cytoskeletal and flagellar microtubules which eliminates the very structures that are used most in protist classification schemes. Molecular methods can generate new characters for use in classification regardless of the morphological complexity of the organism of interest.
The polymerase chain reaction (PCR) amplifies large amounts of a specific DNA sequence from an organism using oligonucleotides complementary to the sequence to prime its replication by the DNA polymerase enzyme. As it theoretically can be used with a single cell as the starting material, this method clearly has many advantages where material is limiting. For a PCR-based method to generate useful data for classification, certain criteria must be met: it must not be dependent on prior knowledge of the genetic organisation of the organism; it must not be affected by the presence of bacterial DNA in the sample, as many eukaryotic microbes are found in association with numerous free-living or symbiotic bacteria; the DNA sequence to be analysed must be ubiquitous and conserved in the organisms of interest; the sequence must at the same time be variable enough so that species specific markers can be identified. Genes that match these apparently contradictory criteria include those encoding the small and large subunit ribosomal RNAs (rDNA).
The structure of rDNA is mosaic in nature, consisting of interspersed stretches of highly conserved, moderately conserved and divergent sequences. Regions can be identified that are conserved among all eukaryotic nuclear rDNAs but are distinct enough from bacterial rDNAs such that no amplification of the latter will occur in the PCR reaction. This method of specifically amplifying eukaryotic rDNA was first described by Medlin et al. [3] as a way to isolate these genes for cloning and DNA sequencing. This approach has been very successful and is largely responsible for the availability of hundreds of the eukaryotic small subunit rDNA sequences currently in the databases. However, DNA sequencing is a labour intensive and expensive proposition if many different related organisms are to be studied. Therefore a way of examining these genes is needed that is comparatively quick and inexpensive, so that multiple isolates of the same species as well as related species can be examined, but that will still yield useful data.
The method I call riboprinting combines the restriction site polymorphism analysis with the rDNA amplification method of Medlin to study sequence variation in the small subunit rDNA indirectly and it was first described by this name in 1991 [4]. The same approach was also developed independently by mycologists [5] and bacteriologists, where it is sometimes known as ARDRA (amplified ribosomal DNA restriction analysis; see [6]). I originally developed the method with the goal of studying relationships among species of Entamoeba, but the same approach has been used successfully in a number of taxonomically diverse protist and fungal genera [7-14].
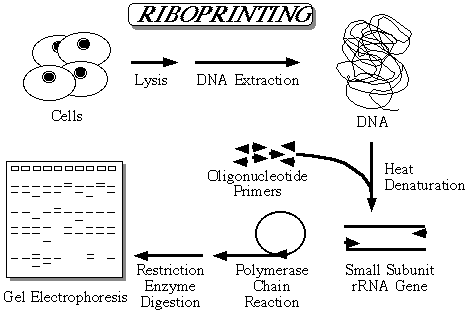
The method is shown diagrammatically in above figure. DNA is isolated from the organisms and used as the template in a polymerase chain reaction. Two of the most highly conserved sequences in the small subunit rDNA are at each end of the small subunit gene. Oligonucleotide primers that are complementary to these regions can are used to amplify the small subunit rDNA. After the PCR is completed, a large quantity of what is essentially the complete small subunit rDNA has been generated. This amplified rDNA is then digested with restriction enzymes and the fragments separated in agarose gels along with a DNA size marker. The gels are then stained and photographed to provide a permanent record. Some of the restriction sites will fall in conserved regions and some in variable regions and this allows sequence polymorphisms to be detected as differences in the restriction fragment sizes.
As can be seen in the figure, species will often share one or more comigrating fragments for each restriction enzyme. Since the number of comigrating fragments will decrease as the gene sequences diverge, riboprinting can also be used to estimate genetic distances among species and algorithms have been developed that allow this to be done [15]. What riboprinting does is sample the gene sequence indirectly. The proportion of the gene sampled depends on the number of restriction enzymes used in the analysis, the size of the restriction enzyme recognition site, and how often they cut the gene. I routinely use 12 enzymes each with a four base recognition sequence and I find that this results in the sampling of 10-15% of the gene sequence without any cloning or sequencing of the DNA being necessary. In addition to generating estimates of genetic distance, the riboprint patterns can be used in parsimony analysis with each DNA fragment being viewed as an individual character.
Riboprinting is thus a method that does not require large amounts of starting material, it is quick, it is reproducible and it uses tools that can be found in almost every molecular biology laboratory.
Polymerase Chain Reaction Amplification
1. Create a standard PCR master mix allowing 45 µl per reaction -
for `Z' reactions thaw the solutions on ice and mix:
0.5xZ µl of each primer in the pair (200 µM stock solution) (Note)
2xZ µl of each dNTP (10 µM stock)
0.5xZ µl of Taq Polymerase
30.5xZ µl of H2O.
Briefly vortex the mix, spin it down in a microcentrifuge and place it on ice.
2. Place 5 µl of each DNA solution in 0.5 ml microcentrifuge tubes on ice, aliquot 45 µl of the master mix into each tube and overlay the reaction mixture with 50 µl of light mineral oil. (Note)
3. Program the thermal cycler to run 30 cycles of: 94° C - 1 minute; 55° C - 1.5 minutes; and 72° C - 2 minutes. Transfer the reaction tubes from the ice to the block and start the cycle sequence.
4. After cycling is over, extract the reactions with 100 µl of CHCl3, briefly spin and remove the supernatant to a clean microcentrifuge tube. Success of the amplification reaction can be monitored by electrophoresis of an aliquot in a 0.8% agarose gel in 1X Tris Borate Buffer at 10 V/cm for 1 hour. The gel is then stained in 1 µg/ml ethidium bromide and destained in water for 15 minutes each on a rotary platform shaker before visualization of the DNA on a UV transilluminator and photographing the gel to obtain a permanent record.
5. Meanwhile, precipitate the amplification products by adding 0.1 volume of 3 M sodium acetate and 2 volumes of 100% ethanol. Vortex the mixture and let it stand for at least 5 minutes at room temperature. Spin the tube at full speed in a microcentrifuge for 15 minutes, wash the pellet with 200 µl of 70% ethanol by spinning for 5 minutes, then dry and resuspend it in 50 µl H2O.
6. To visualize the full restriction pattern it is necessary to remove residual primers by passing the amplification product over a Sephacryl S400 (1 ml packed volume) spin column before digestion (see here for details).
7. The product is digested with restriction enzymes. The more enzymes used the more information is obtained - I recommend 12 enzymes all of which recognise a 4-base sequence.
Riboprinting has its limitations:
1. Since only 10-15% of the gene sequence is sampled by this indirect method, significant variation can be present and will go undetected unless divergence falls within restriction sites. Similarly, if two restriction sites fall close together the resulting fragment will not be visible in the agarose gels and, if the sites are very close, the corresponding reduction in size of the adjacent restriction fragment may be too small to be noticed. Undoubtedly, riboprinting will miss some variation.
2. It is known that some species defined by reproductive isolation have small subunit rDNAs that are identical in sequence [16]. Therefore, identical riboprints do not indicate that the organisms necessarily belong to the same species.
3. Building trees based on fragment co-migration data assumes that all comigrating fragments are homologous. This will usually but not always be the case. Fragment co-migration can only be used to estimate relationships with any accuracy when the organisms are quite closely related [17]. Converting riboprints into restriction maps and using the number of shared restriction sites to calculate genetic distances is known to provide more accurate estimates of relatedness.
4. Riboprinting is much less sensitive at detecting variation than isoenzyme analysis, for example. Isoenzyme variation is commonly seen among organisms that have identical riboprints. Restriction enzyme fingerprinting of less conserved genes may be informative in such cases.
5. If a mixture of small subunit rDNA sequence variants coexist in the same organism riboprinting may or may not reveal this fact. Where two forms of the gene exist differing at a single nucleotide position that coincides with a restriction site the resulting pattern will likely be interpreted as resulting from incomplete restriction enzyme digestion of the PCR product even if both variants make up a significant proportion of the gene complement. One such case has been identified, but only because a complete gene sequence was already available [18]. Minor variants may not be noticed at all.
6. Occasionally, related species will differ by the presence of an intron in the small subunit rDNA of one but not the other [19]. Such a situation will be easily recognized as the small subunit rDNA amplification products will differ in size by several hundred basepairs. The presence of introns, while interesting, makes comparison of riboprint patterns for phylogenetic analysis difficult at best unless they are present in in all isolates being compared.
7. Mixtures of organisms will produce difficulty in interpretation of restriction digest patterns. In some cases their existence will be obvious from the presence of two or more bands in the undigested product gel patterns. However, this will not always be the case. Mixtures will give rise to patterns where the sum of the fragment sizes exceeds the size of the amplification product.
As long as these limitations are recognized, riboprinting is a fast, reproducible way to obtain useful data from small numbers of organisms. It has the ability to detect cryptic genetic variation, to identify morphologically conservative organisms, to uncover misidentified organisms and culture mix-ups, to act as an independent check on DNA sequencing accuracy, and to provide data that can be used to estimate relatedness and to generate phylogenetic trees.
Note 1: The choice of the primer pair used depends on the species involved. Amplification of the SSU-rDNA of many species uses the primer pair RD5 and RD3 which amplifies almost the entire SSU-rRNA gene. Amplification of the SSU-rDNA of trichomonads uses the primer pair TRD5 and TRD3 [20], while diplomonads and microsporidia would also require group specific primers due to sequence divergence in the primer binding regions of the genes. Identification of all species then relies on digestion of the amplification product with a battery of restriction enzymes.(Back)
Note 2: It is often useful to try at least two DNA concentrations for each DNA preparation, eg. a 1:10 and a 1:100 dilution. Include a negative control using 5 µl H2O instead of DNA.(Back)
1. Clark, C. G. 1993. PCR detection of pathogenic Entamoeba histolytica and differentiation from other intestinal protozoa by riboprinting. In: Persing, D. H., Smith, T. F., Tenover, F. C. & White, T. J. (ed.), Diagnostic molecular microbiology. Principles and applications. ASM Press, Washington, D.C. Pp. 468--474. (Back)
2. Clark, C.G. 1992 Riboprinting: a molecular approach to the identification and taxonomy of protozoa. in Protocols in Protozoology, Volume 1.(J.J. Lee, A.T. Soldo, eds) Allen Press, Lawrence, Kansas. D-4.1 -- D-4.4. (Back)
3. Medlin, L., Elwood, H. J., Stickel, S. & Sogin, M.L. 1988. The characterization of enzymatically amplified eukaryotic 16S-like rRNA coding regions. Gene 71:491--499. (Back)
4. Clark, C. G. & Diamond, L. S. 1991. The Laredo strain and other `Entamoeba histolytica -like' amoebae are Entamoeba moshkovskii. Mol. Biochem. Parasitol. 46:11--18. (Back)
5. Vilgalys, R. & Hester, M. 1990. Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172:4238--4246. (Back)
6. Vaneechoutte, M., Rossau, R., De Vos, P., Gillis, M., Janssens, D., Paepe, N., De Rouck, A., Fiers, T., Claeys, G. & Kersters, K. 1992. Rapid identification of bacteria of the Comamonadaceae with amplified ribosomal DNA-restriction analysis (ARDRA). FEMS Microbiol. Letts. 93:227--233. (Back)
7. Clark, C. G. 1997. Extensive genetic diversity in Blastocystis hominis. Mol. Biochem. Parasitol. 87: 79-83. (Back)
8. Clark, C. G. & Pung, O. J. 1994. Host-specificity of ribosomal DNA variation in sylvatic Trypanosoma cruzi from North America. Mol. Biochem. Parasitol. 66:175--179. (Back)
9. Clark, C. G., Martin, D. S. & Diamond, L. S. 1995. Phylogenetic relationships among anuran trypanosomes as revealed by riboprinting. J. Euk. Microbiol. 42:92--96. (Back)
10. De Jonckheere, J. F. 1994. Riboprinting of Naegleria spp.: small-subunit versus large subunit rDNA. Parasitol. Res. 80:230--234. (Back)
11. Jerome, C. A. & Lynn, D. H. 1996. Identifying and distinguishing sibling species in the Tetrahymena pyriformis complex (Ciliophora, Oligohymenophorea) using PCR/RFLP analysis of nuclear ribosomal DNA. J. Euk. Microbiol. 43:492--497. (Back)
12. Molina, F. I., Inouye, T. & Jong, S.- C. 1992. Ribosomal DNA restriction analysis reveals genetic heterogeneity in Saccharomyces cerevisiae Meyen ex Hansen. Int. J. Syst. Bacteriol. 42:499--502. (Back)
13. Molina, F. I., Inouye, T. & Jong, S.- C. 1992. Determination of infraspecific relationships in Kluyveromyces marxianus by riboprinting. Mycotaxon 43:49--60. (Back)
14. Brown, S. & De Jonckheere, J. F. 1994. Identification and phylogenetic relationships of Vahlkampfia spp. (free-living amebas) by riboprinting. FEMS Microbiol. Letts. 115:241--246. (Back)
15. Nei, M. & Li, W.- H. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76:5269--5273. (Back)
16. Sogin, M. L., Ingold, A., Karlok, M., Nielsen, H. & Engberg, J. 1986. Phylogenetic evidence for the acquisition of ribosomal RNA introns subsequent to the divergence of some of the major Tetrahymena groups. EMBO J. 5:3625--3630. (Back)
17. Nei, M. 1987. Molecular evolutionary genetics. Columbia University Press, New York, NY. (Back)
18. Clark, C. G. & Diamond, L. S. 1997. Intraspecific variation and phylogenetic relationships in the genus Entamoeba as revealed by riboprinting. J. Euk. Microbiol. 44:142-154. (Back)
19. De Jonckheere, J. F. & Brown, S. 1994. Loss of the ORF in the SSUrDNA group I intron of one Naegleria lineage. Nucl. Acids Res. 22:3925--3927. (Back)
20. Silberman, J.D., Clark, C.G., Sogin, M.L. 1996 Dientamoeba fragilis shares a recent common evolutionary history with the trichomonads. Mol. Biochem. Parasitol. 76: 311--314. (Back)
21. Clark, C.G. 1997 Riboprinting: a general tool for the study of genetic diversity in microorganisms. J. Euk. Microbiol. 44: 277--283. (Back)